Modelling metal transport in the deep Earth
Metals are transported from mantle or lower crust to shallow crust via various kinds of chemical compositions then form economic ores. Melt-brine-vapor systems cover some types of ore deposits with economic importance such as porphyry, skarn and epithermal deposits. Reliable knowledge of metal behavior in melt-brine-vapor system is essential for understanding the enrichment, transport and precipitation of metals and the formation of ore deposits. The aim of this study is to provide molecular-level understanding of metal in melt-brine-vapor systems in the deep earth to address key fundamental questions about the origins, enrichments, transports and deposits of economic valuable metals in the global elements cycling. This fundamental knowledge has significant implications for mineral exploration and for maintaining a sustainable rate of discovery of ore deposits.

Area of science
Geosciences
Systems used
Magnus, Managed Storage and Visualisation
Applications used
The Challenge
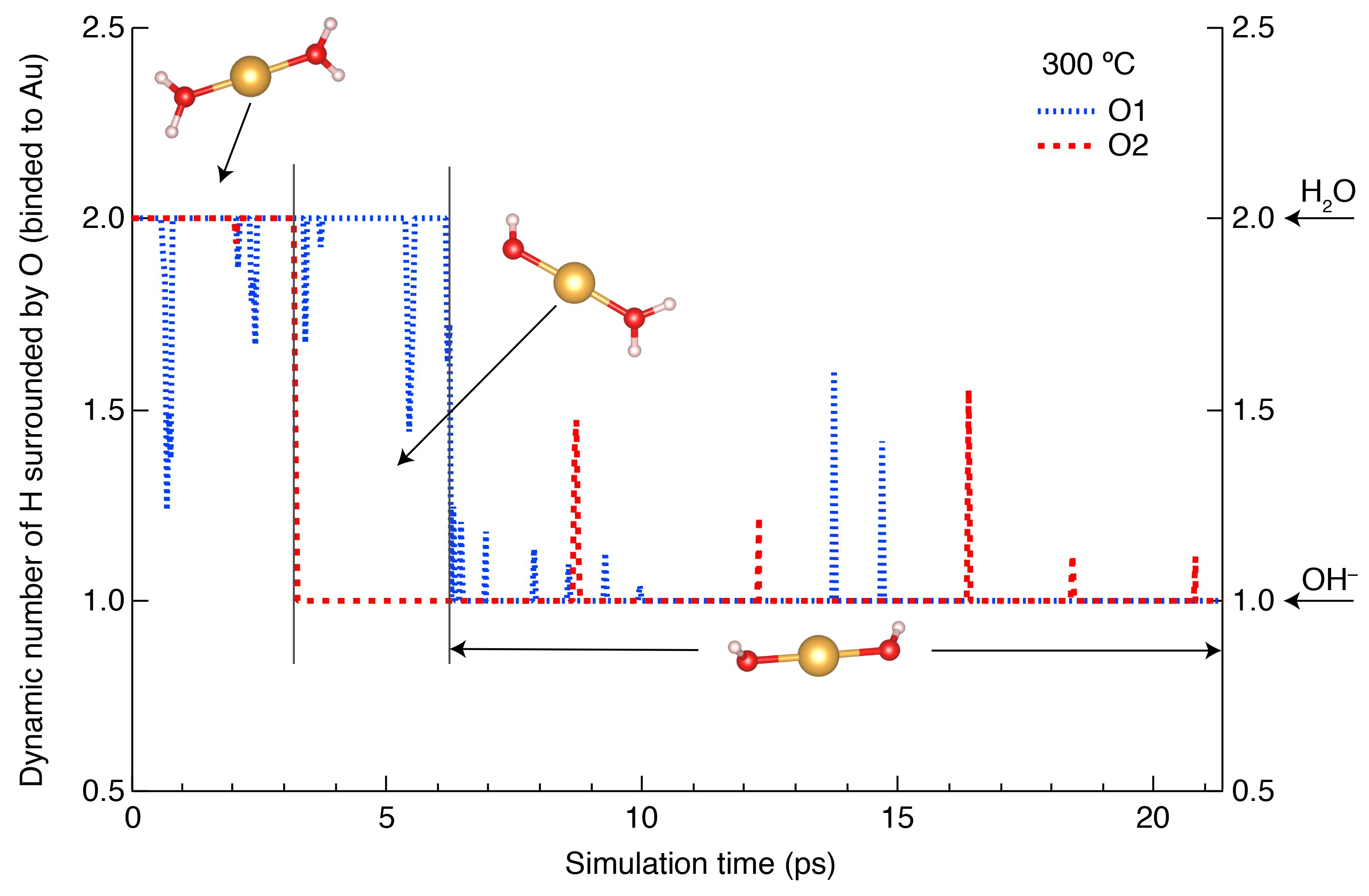
MD simulations use fundamental physical laws to calculate the atomic momenta and positions of all the atoms in the modelled system through time, providing a detailed picture of the structure and dynamics of solvent-complex interactions. When rigorously calibrated against experimental data, MD simulations can provide a realistic estimate of chemical speciation and reactions at extreme temperatures, pressures and compositions where experiments are challenging or impossible.
The Solution
In this project, we use ab initio MD to investigate the speciation and thermodynamic properties of selected metals and ligands at high T-P conditions, then we apply large-scale force field MD in vapor-liquid multi-phase systems under hydrothermal conditions to calculate the metal partitioning between different phases. The results of complexation and partitioning coefficients can be compared directly to experiments. This project leverages these new capabilities to address fundamental questions about the partitioning of metals in melt-brine-vapor systems and the role of phase separation in the global metal cycling.
The Outcome
Research highlights and achievements (2018 onwards):
We acknowledge Pawsey for the HPC support to the follow research work:
Peer-reviewed journal papers
Mei, Y., Liu, W., Brugger, J., & Guan, Q. (2020). Gold solubility in alkaline and ammonia-rich hydrothermal fluids: insights from ab initio molecular dynamics simulations. Geochimica et Cosmochimica Acta. doi:https://doi.org/10.1016/j.gca.2019.12.031
2 Syverson, D. D., Etschmann, B., Liu, W., Ram, R., Mei, Y., Lanzirotti, T., . . . Brugger, J. (2019). Oxidation state and coordination environment of Pb in U-bearing minerals. Geochimica et Cosmochimica Acta, 265, 109-131. doi: https://doi.org/10.1016/j.gca.2019.08.039
3 Etschmann, B., Liu, W., Mayanovic, R., Mei, Y., Heald, S., Gordon, R., & Brugger, J. (2019). Zinc transport in hydrothermal fluids: On the roles of pressure and sulfur vs. chlorine complexing. American Mineralogist, 104(1), 158-161. doi:http://dx.doi.org/10.2138/am-2019-6719
4 Mei, Y.*, Liu, W., Migdiov, A. A., Brugger, J. & Williams-Jones, A. E. (2018). CuCl Complexation in the Vapor Phase: Insights from Ab Initio Molecular Dynamics Simulations. Geofluids, 2018, 12. doi:http://doi.org/10.1155/2018/4279124
5 Mei, Y.*, Liu, W., Brugger, J., Sherman, D. M., & Gale, J. D. (2018). The dissociation mechanism and thermodynamic properties of HCl(aq) in hydrothermal fluids (to 700 °C, 60 kbar) by ab initio molecular dynamics simulations. Geochimica et Cosmochimica Acta, 226, 84-106. doi:https://doi.org/10.1016/j.gca.2018.01.017
6 Etschmann, B. E., Mei, Y., Liu, W., Sherman, D., Testemale, D., Müller, H., . . . Brugger, J. (2018). The role of Pb(II) complexes in hydrothermal mass transfer: An X-ray absorption spectroscopic study. Chemical Geology. doi:https://doi.org/10.1016/j.chemgeo.2018.10.022
In revision:
Qiushi Guan, Yuan Mei, Barbara Etschmann, Denis Testemale, Marion Louvel, Joël Brugger, Yttrium complexation and hydration in chloride-rich hydrothermal fluids: insights from ab initio molecular dynamics simulations and X-ray absorption spectroscopy, submitted to Geochimica et Cosmochimica Acta