Understanding cement hydration and phase diagram stability using first-principles
Cement-based materials (CBM) are playing a fundamental role in the economic development in Australia, especially in Western Australia. For its application, very large amounts of cement clinker is produced. To minimise the environmental impact, we need a better understanding of the structural and electronic properties of cement phases, as well as their interaction with water. Also, the phase diagram stability of cement raw materials needs to be investigated so that new ways for cement production with less energy consumption may be found. First-principle calculations are a promising approach for investigating above topics. The main objective of this study is as follows: (i) understanding the structural and electronic properties of cement phases; (2) investigating the surface characteristics and its interaction with water molecules; (3) understanding the phase diagram stability of raw materials. Such a study is vital for the sustainable development of the cement industry.

Area of science
Chemical Sciences, Environmental Sciences, Geosciences, Physical Sciences
Systems used
Magnus
Applications used
VASP 5.4.4The Challenge
The application of cement-based materials (CBM) plays a fundamental role in the economic development in Australia, especially in Western Australia where the importance of the mining industry is pivotal to the state’s economy. In civil engineering, concrete is a type of CBM made of fine to coarse aggregate, cement and mixing water. It is the most widely used construction material internationally, with an annual production exceeding ten cubic kilometres worldwide. In Australia, a massive demand surge for concrete is expected over the next five years. Approximately $75 billion has been allocated to infrastructure spending over the next ten years in Australia for new airports, tunnels, bridges etc. In mining engineering, cemented paste backfill (CPB) is another type of CBM widely utilised. As one of Australia’s ‘five pillar economy’, the mining industry is introducing CPB technology for its technical, economic and environmental benefits.
This study will promote understanding of cement hydration process and the phase diagram stability of cement raw materials. Such investigation could suggests new ways to achieve the required mechanical properties of CBM, but with reduced cement consumption, in order to minimise environmental impact. Also, a more energy-efficient cement production method could be proposed to reduce the CO2 footprint during cement production.
The findings of this research will benefit society considering that CBM plays a vital role in many aspects of science, technology and engineering today. The expanding demand for CBM in an era with increasing environmental concerns will require fundamental innovations in the cement industry. Thus, implementation of the recommended approach derived from the results of this research will enable cement producers to reduce their energy consumption. The construction industry will be guided on how to control cement hydration to meet specific engineering requirements in various cases. For the mining industry implementing CPB technology, this research will help the achievement of equivalent mechanical properties of CPB materials, but with less cement consumption. Therefore, a better understanding of the structural and electronic properties of cement phases, its interaction with water and the phase diagram stability during cement clinker production must be achieved by first-principles calculations.

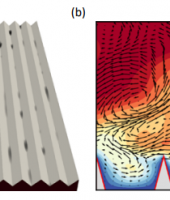
Figure. 3 Influence of dissociative water adsorption on structural and Bader charge of β-C2S (011) surface: (a) structural change, (b) Bader charge change, and (c) atomic labelling
The Solution
All first-principles calculations will be performed using the VASP package. Different exchange-correlation functionals will be tested for cement phases to select the most appropriate one for each task. A convergence test will be followed to determine the energy cutoff and the k-mesh.
Firstly, the structural and electronic properties for each cement phase will be investigated. By comparing with some results from the literature, the computational accuracy can be verified. Also, the influence of ion impurity and defects on the structural and electronic characteristics of bulk materials can be investigated. The next step will be the surface investigation, including surface reconstruction, structural properties of surface and surface energy. The adsorption of water molecules will be a further step to investigate the interaction of water molecules with cement surfaces. Water adsorption with different coverage can be investigated, together with its adsorption on defected surfaces. Elastic band method will also employed for transit state searching. Phase diagram stability will be investigated by density-functional perturbation theory simultaneously with the objective to find new ways to reduce the cement production temperature.
This project consists of many individual tasks, such as unit cell optimisation and surface reconstruction. Approximate 100-200 cores will be used for each individual task and tens of individual tasks can be run simultaneously. The exact number of cores and simultaneous tasks will be determined during the project. Pre/post processing can be performed using desktops after the calculation results are downloaded. To carry out the whole project, 1,382,400 core hours are needed. As some VASP output files are large in size (up to 1GB), 1 TB memory are expected for this project.
The Outcome
All DFT calculations were conducted using the computational resources provided by this project. Our research cannot be proceeded without the provided core hours.
With the core hours in the current project, the single water adsorption on all low-index surfaces of β-C2S and M3-C3S was investigated using DFT-D calculations. The surface energy was calculated and the influence of surface cleavage discussed. Both dissociative and molecular adsorptions were investigated. The results show that the adsorption energy ranged from -1.210 eV to -0.752 eV on β-C2S surfaces and ranged from -3.242 eV to -0.732 eV on M3-C3S surfaces. Molecular adsorption was energetically favoured on β-C2S surfaces while dissociative adsorption was energetically favoured on M3-C3S surfaces. A Wulff construction was used to describe the equilibrium morphology with and without an adsorbed water molecule for the β-C2S phase. Water adsorption promoted the solid dissolution by weakening the bond among surface atoms. Electron transfer was observed mainly from the surface atoms to water atoms during the adsorption. These findings provide a novel insight into the adsorption of a water molecule on different calcium silicate surfaces using the same level of theory, which includes a representation of van der Waals forces, therefore, laying the foundation for better understanding the hydration mechanism.
List of Publications from this project
Chongchong Qi, Dino Spagnoli, Andy Fourie. DFT-D study of single water adsorption on low-index surfaces of calcium silicate phases in cement (2020). Applied Surface Science. 518, 146255.