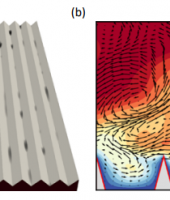
Atomistic simulation of minerals, materials and geochemistry
Minerals are of great significance to everything from health, to the environment and economy. Despite their ubiquitous importance, our understanding of how minerals form remains incomplete. This in turn limits our ability to control and manipulate their formation. Being able to exert such control would have benefits in reducing scale production in industrial processes through to processes such as long term carbon sequestration. Our research focuses on the family of biominerals that are pivotal to both these aforementioned areas, as well as responsible for the minerals that occur in the human body. The objective of our project is to understand how such minerals assemble at the atomic level – something that is hard to achieve in the laboratory, but is becoming possible using computer models

Area of science
Geochemistry, Physical chemistry
Systems used
•Magnus •Zeus •Nimbus •Topaz
Applications used
LAMMPS, OpenMM, CP2K, CRYSTAL, CASTEP, PSI4The Challenge
Crystals are the basis of much of the solid matter around us. Understanding how they form from their component molecules and/or ions in solution has long been a challenge. If we were able to control this process then we would be able to limit the formation of scale that blocks pipes and vessels that costs industry in lost productivity, as well as being able to design new materials such as pharmaceuticals. In the context of minerals specifically, the formation of crystals is one of the processes by which systems are purified in industry and in nature this goes to form everything from sand on the beach, to teeth. At the moment we have models of the rate of crystal growth based on fitting macroscopic experimental data. However this requires many assumptions about the component steps that are happening. Even worse, there is even less known regarding the initial “birth” of crystals, referred to as nucleation
The Solution
One way that we can determine the individual steps take by ions and molecules to form a crystal is through using computer simulation. Here we can model what happens when atoms are placed in a given arrangement and how this changes with time through the use of a technique known as molecular dynamics. Unfortunately the amount of real time that can observed in this way is limited to typically less than a millionth of a second, which is slower than most experiments, especially geological processes. However, by applying a bias to the simulations that allows them to jump over large barriers we can dramatically accelerate the evolution of the model. Although simple knowledge of time is lost, we gain a wealth of information regarding the thermodynamics, which is the driving force for chemical and physical reactions. Nevertheless, the computational demands remain high as billions of steps are still required – something that is only possible with state of the art supercomputers.
The Outcome
Although we know how to overcome the problem of slow processes in our simulation model, the process still has to be solved in discrete time steps that are of the order of 10^-15 of a second due to the fundamental nature of chemical interactions. Therefore each model requires up to a billion iterations to acquire sufficient data to be useful. Each iteration can also be expensive when using the most accurate methods available. Only through the use of many thousands of processors working simultaneously on the same model is this feasible. Access to world-class supercomputers, such as the Magnus system with tens of thousands of cores, is therefore critical to our research. In future, new technologies, such as graphical processing units, may also allow us to speed up the evaluation of each step – something that we have also been exploring using the resources of the Pawsey Centre.
List of Publications
1. Q. Lu, T. Willhammar, B.-B. Sun, N. Hedin, J.D. Gale, D. Gebauer, 2020. “Introducing the crystalline phase of dicalcium phosphate monohydrate”, Nature Communications, 11, 1546.
2. Raiteri, P., A. Schuitemaker, J.D. Gale, 2020. “Ion pairing and multiple ion binding in calcium carbonate solutions based on a polarizable AMOEBA force field and ab initio molecular dynamics“, Journal of Physical Chemistry B, 124, 3568-3582.
3. Li, C., A.G. Shtukenberg, L. Vogt-Maranto, E. Efrati, P. Raiteri, J.D. Gale, A.L. Rohl and B. Kahr, 2020. “Why are some crystals straight?”, Journal of Physical Chemistry C, 124, 15616-15624.
4. Brugman, S.J.T, P. Raiteri, P. Accordini, F. Megens, J.D. Gale and E. Vlieg, 2020. “Calcite (104) surface-electrolyte structure: A 3D comparison of surface X-ray diffraction and simulations“, Journal of Physical Chemistry C, 124, 18564-18575.
5. Krauss, P., 2020 “Basis set extrapolations for density functional theory“, J. Chem. Theory Comput., 16, 5712-5722.
6. Demichelis, R. 2020. “The structure of uric acid dihydrate crystals revisited via first-principle methods“, Australian J. Chem., 73, 923-928.
7. Raiteri, P., P. Kraus and J.D. Gale, 2020. “Molecular dynamics simulations of liquid-liquid interfaces in an electric field: the water-1,2-dichloroethane interface”, J. Chem. Phys., 153, 164714.
8. Aufort, J. and R. Demichelis, 2020. “Magnesium impurities decide the structure of calcium carbonate hemihydrate“, Cryst. Growth Des., 20, 8028-8038.
9. Lunkov, S.A., J.D. Gale and K.R. Barnard, 2020. “A quantum chemical study of LIX63 hydroxyoxime syn/anti isomerisation“, Mol. Simul., 46, 1530-1541