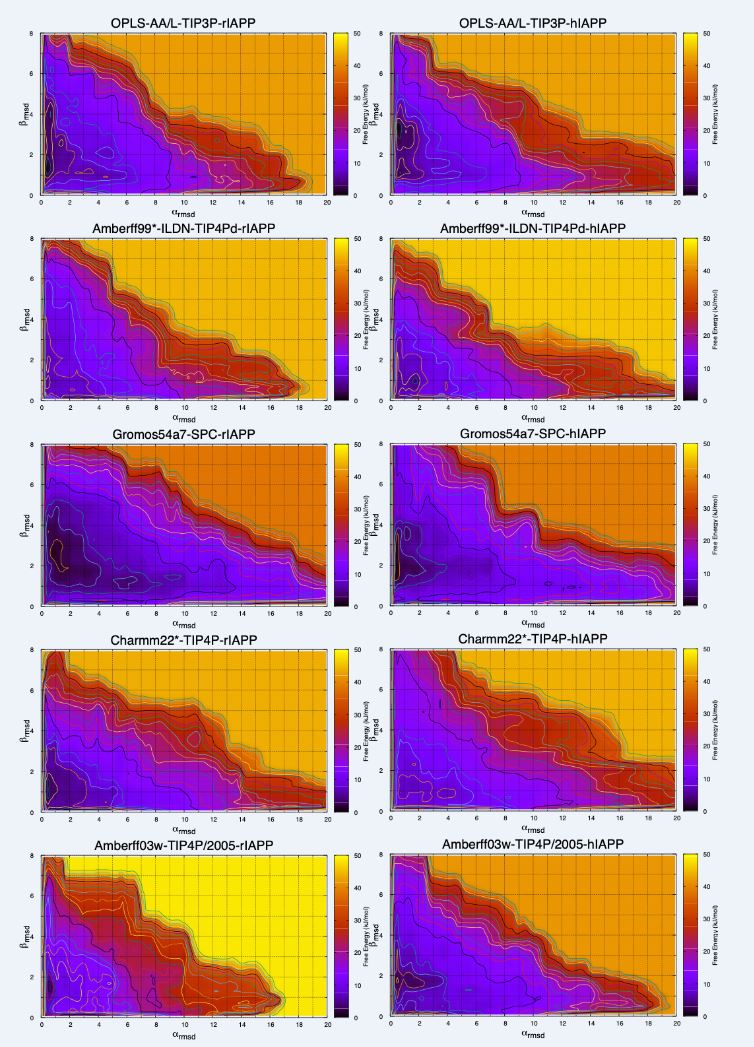
Molecular Dynamics Simulation of Biophysical Phenomena
This project aims to use large scale computer simulation methods to describe the behaviour of a range of molecular systems of biological interest, such as proteins and cell membranes. These biomolecular systems have importance for our understanding of how (1) proteins deleteriously misfold and aggregate in neurodegenerative states, (2) sugars and other cryoprotective agents affect cell membranes, (3) sugars and nucleic acids likely stabilised protocells in the origins of life, (4) therapeutic and venom peptides penetrate and/or damage cell membranes, and (5) proteins can be detected in electrochemical devices. State-of-the-art computational methods will be used to study in atomistic detail how biomolecules interact with each other in normal, artificial and disease states, which will help in the design of better biotechnological and pharmaceutical approaches to improve health outcomes.

Area of science
Biological Sciences, Computational biophysics, Geosciences
Systems used
Magnus
Applications used
Gromacs, AMBERThe Challenge
Biological events at the molecular level occur at relatively large length and time scales. Understanding how these events occur in detail requires the use of sophisticated experimental and computational methods. In this project in particular the key challenges are to characterise (1) the mechanism by which proteins involved in neurodegenerative diseases (such as Alzheimer’s) fold incorrectly and form large aggregates, (2) the changes to the structure and function of model cell membranes when exposed to the deleterious action of cryosolvents, (3) the molecular mechanisms that led to the formation and stabilisation of early protocells in the origins of life, (4) the molecular mechanism by which venom peptides interact and damage cell membranes, and (5) the mechanism by which proteins adsorb and change their structure at liquid-liquid interfaces in electrochemical sensors.
The Solution
State-of-the-art molecular simulation methods coupled with high performance computing facilities allow the study of biological processes in exquisite atomistic detail in order to elucidate their molecular mechanisms.
The Outcome
High performance computing is essential for the simulation of complex biological systems in the time and length scales necessary to characterise molecular processes. The molecular complexity of these systems, the complexity of the algorithms used to investigate them, and the large amounts of data generated mean that only through access to supercomputing facilities can these problems be tackled.
List of Publications
(a) Stachura, S., Malajczuk, C.J. Kuprusevicius, E. & Mancera, R.L. “Influence of bilayer size and number in multi-bilayer DOPC simulations at full and low hydration”, Langmuir, 2019, vol. 35, pp. 2399-2411.
(b) Booth, S.G., Felisilda, B.M.B., Alvarez de Eulate, E., Gustafsson, O.J.R., Arooj, M., Mancera, R.L., Dryfe, R.A.W., Hackett, M.J & Arrigan, D.W.M., “Secondary structural changes in proteins as a result of electroadsorption at aqueous-organogel interfaces”, Langmuir, 2019, vol. 36, pp. 5821-5829.
(c) Arooj, M., Arrigan, D.W.M. & Mancera, R.L. “Characterisation of the protein-facilitated ion-transfer mechanism at a polarised aqueous-organic interface”, Journal of Physical Chemistry B, 2019, vol. 123, pp. 7436-7444.
(d) Kompella, V.P.S., Stansfield I., Romano M.C. & Mancera, R.L. “Definition of the minimal contents for the molecular simulation of the yeast cytoplasm”, 2019, Frontiers in Molecular Biosciences, vol. 6, article 97.
(e) Stachura, S.S., Malajczuk, C.J. & Mancera R.L. “Does sucrose change its mechanism of stabilization of lipid bilayers during desiccation? The influence of hydration and concentration”, 2019, Langmuir, vol. 35, pp. 15389-15400.
(f) Deplazes, E., Chin, Y.K-Y., King, G.F. & Mancera, R.L. “The unusual geometry of cross-strand disulfides is critical to the stability of small β-hairpin peptides: the case of the spider peptide Gomesin”, 2020, Proteins: Structure, Function and Bioinformatics, vol. 88, pp. 485-502.
(g) Martinotti, C., Ruiz-Perez, L., Deplazes, E. & Mancera, R.L. “Molecular dynamics simulation of the interaction of small molecules with biological membranes”, ChemPhysChem, 2020, accepted for publication on 20 May 2020.